
CMEDIA: Scientists have known for years that amyloid fibrils—fibrous, ropelike structures formed by closely linked protein molecules—are present in the brains of patients with Alzheimer’s and Parkinson’s diseases and likely play a role in the progression of these disorders.
Now, UCLA biochemists have discovered such fibrils in the brains of people with a form of frontotemporal lobar degeneration, or FTLD, the most common neurodegenerative disorder after Alzheimer’s and Parkinson’s. But surprisingly, the type of protein they expected to find in these potentially harmful fibrils wasn’t there at all; instead, a little-known protein called TMEM106B was identified as the culprit.
The findings, published in the journal Nature, are likely to lead to a new focus on TMEM106B in FTLD and similar brain diseases, the researchers said.
FTLD, which involves damage to the brain’s frontal and temporal lobes, strikes people earlier in life than Alzheimer’s and Parkinson’s, causing dementia in 80 out of 100,000 people between the ages of 45 and 64. Symptoms can include acute changes in behavior and a decline in language skills. The form of the disease studied by the researchers is characterized molecularly by dense, spherical aggregates in brain cells, which are made up of the protein TDP-43.
The UCLA team presumed that if there were amyloid fibrils in the brains of people with FTLD, these fibrils would be composed of TDP-43.
The study’s lead authors, UCLA molecular biology graduate student Yi Xiao (Sean) Jiang and UCLA postdoctoral fellow Qin Cao, both working in the laboratory of UCLA professor David Eisenberg, were indeed able to extract amyloid fibrils from frozen brain tissues provided by the Mayo Clinic from four deceased patients who had been diagnosed with FTLD-TDP.
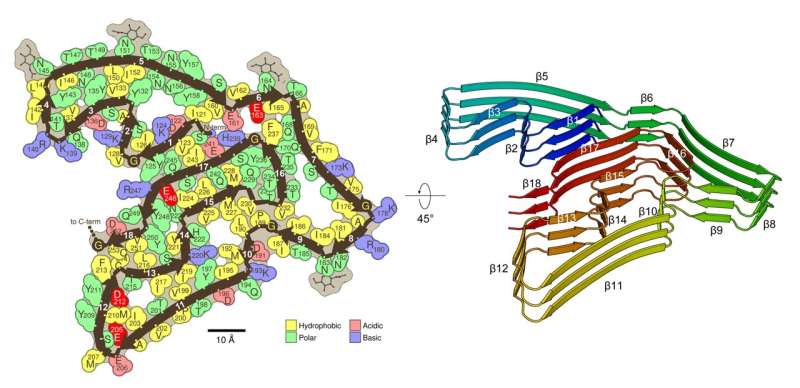
However, using a technique called cryogenic electron microscopy, or cryo-EM—which allowed them to image large biomolecules in extraordinary detail—they, along with UCLA bioinformatics researcher Michael Sawaya, determined that the fibrils were composed solely of TMEM106B, or transmembrane protein 106B.
Not much is known about TMEM106B, although genetic scientists found evidence a decade ago that a mutation of the protein is a risk factor for FTLD, said senior author Eisenberg, the Paul D. Boyer Professor of Molecular Biology at UCLA and a Howard Hughes Medical Institute investigator.
Like a golf course: The structure of amyloid fibrils in FTLD
Pathological deposits of amyloid proteins are associated with more than 50 degenerative and potentially fatal diseases, according to Eisenberg, who has studied amyloid fibrils for years.
In a 2005 Nature paper, Eisenberg and an international team of chemists and molecular biologists reported that these fibrils are made up of proteins that interlock like the teeth of a zipper—an observation that has been supported by recent studies. TMEM106B, he said, forms multiple molecular zippers.
The team’s structural analysis of TMEM106B fibrils shows that they share features with the pathogenic fibrils seen in Alzheimer’s and Parkinson’s but are more complex, Eisenberg noted. Like those other fibrils, they are made of stacks of thousands of layers, each layer formed by single protein molecules with straight segments and bent corners, folding into a complex shape.
In TMEM106B, the folded protein chain has 18 straight segments, which the researchers liken to the 18 fairways of a golf course—and like a golf course, the first and 18th fairways are near each other. “So we say TMEM106B has a golf course–like fold,” Eisenberg said.
Whether the TMEM106B amyloid fibrils contribute to causing FTLD-TDP remains to be seen. It is also unclear what, if any, role may be played by the TDP-43 proteins, whose normal function is to shepherd RNA molecules carrying DNA blueprints for proteins from the nucleus of brain cells to the cytoplasm.
“TMEM106B may be found to be a cause of FTLD. In that case, our knowledge of the structure will aid in the design of therapeutics,” Eisenberg said. “Further research may also discover a connection between the actions of TMEM106B and TDP-43. It’s too early to tell.
“But at the least, the present paper will alert the community of researchers studying neurodegeneration that a new protein may potentially play a role,” he said.
UCLA co-authors of the paper are Romany Abskharon, Peng Ge, Janine Fu, Rachel Ogorzalek and Joseph Loo. Michael DeTure and Dennis Dickson of the Mayo Clinic in Jacksonville, Florida, provided patient tissues and contributed neuropathological analyses.
Lead author Cao now heads his own lab at China’s Shanghai Jiao Tong University.


